

こんにちは、製薬研究者のMasuです。
マイクロアレイやプロテオミクスなど遺伝子やタンパク質のデータ(以下遺伝子とします)を大量に扱うとき、もしくは気になっているタンパク質のリストがあるとき、各遺伝子の機能や局在データを一つ一つ手作業で集めることは困難です。
それでもこれが研究だからしょうがない、、と割り切って一つ一つ探すのも一つの手ですが、pythonを使うと超効率的に情報収集ができます。
他のサイトでもwebスクイピングのコードは記載されていますが、それらは実用的でないことがほとんどです。
なぜなら我々はエクセルで作成したリストを使いたい場合が多いからです。
そこで今回は、タンパク質/遺伝子データベースであるUniprotを情報源に、エクセルで作成されたリストから遺伝子・タンパク質の情報を超効率的に収集する方法について紹介したいと思います。
用意するもの
以下を用意しましょう。
- python
- エクセルで作成したタンパク質のリスト
python
pythonを使える環境がまだ無い方は、以下の記事を参考にまずはpythonをインストールしてください。

エクセルで作成したタンパク質のリスト
最も理想なのは、以下のようなuniprotKB accession numberで作成されたリストです。
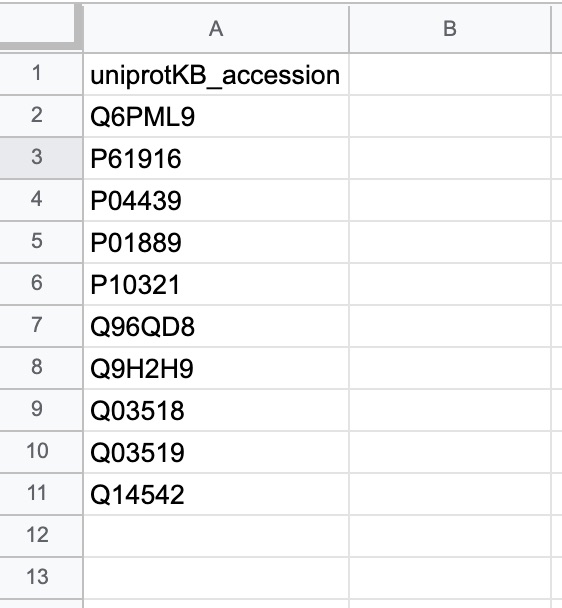
uniprotKB accession numberとは、uniprotで検索した際、以下の赤字で囲った部分で示されるIDのことです。
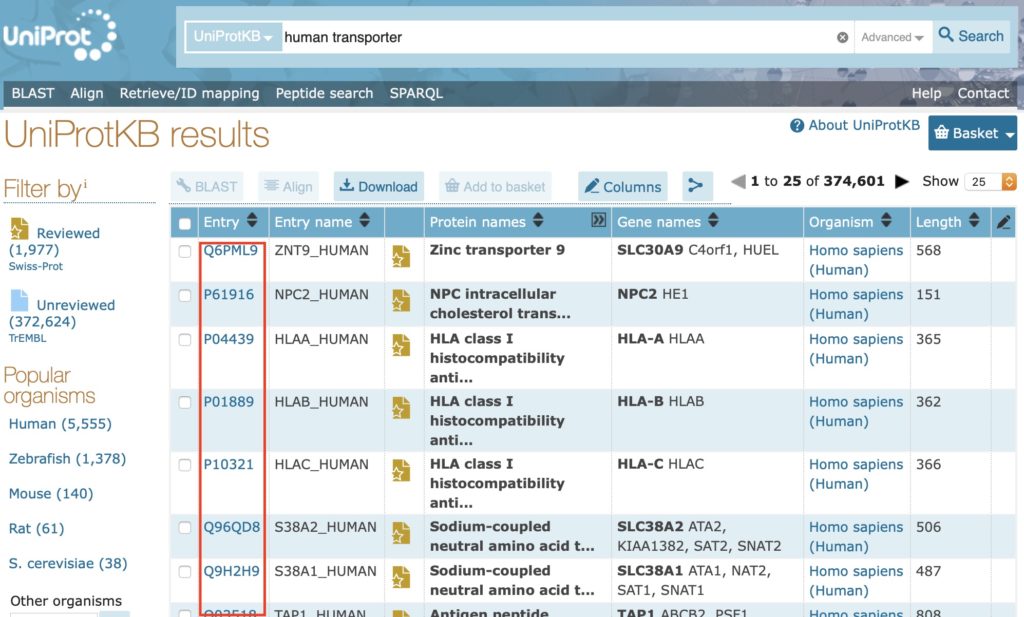
ですがもしタンパク質・遺伝子のリストがuniprot accession IDで作成されていないのであれば、uniprotが提供するID変換ツールを以下を参考にして使用してください。
【ID変換】uniprot ID Mapping (必要な場合のみ)
ID変換ツールとしてuniprot ID Mappingを使用します。
(例)Gene nameからuniprotKB accession IDへの変換
①まず赤枠で囲った部分に変換したいIDのリストを貼り付けてください。
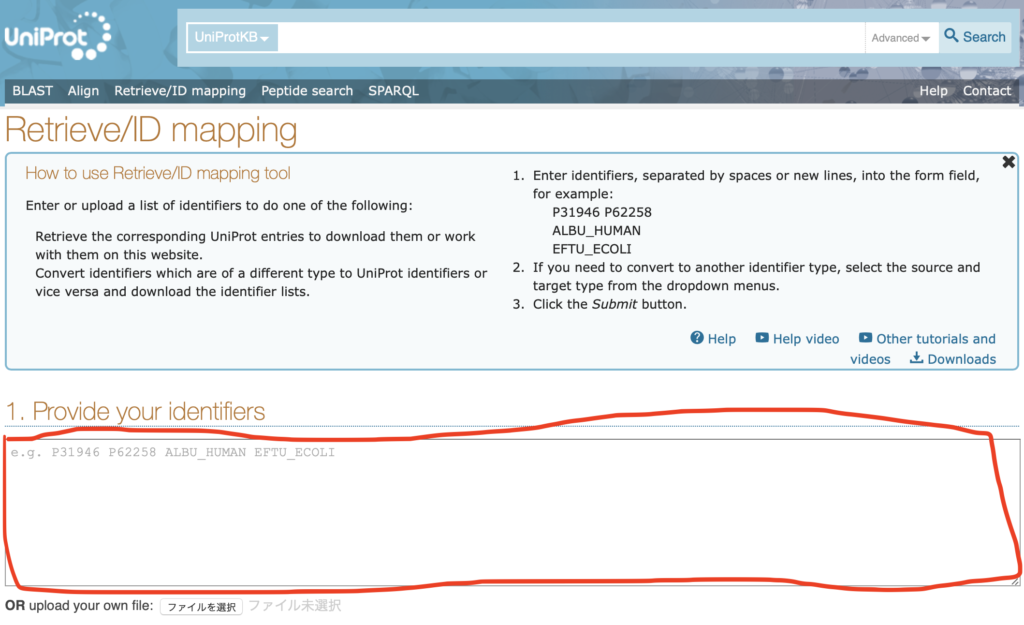
②貼り付けたら、FromをGene nameに、ToをUniprotKBに変えて下さい。別のIDから変換したい場合は、Fromの内容を適宜変えてください。またGene nameの時はOrganismも選択する必要があるようです。ここではhumanと打ち込んでみてください。候補リストが出てくると思います。
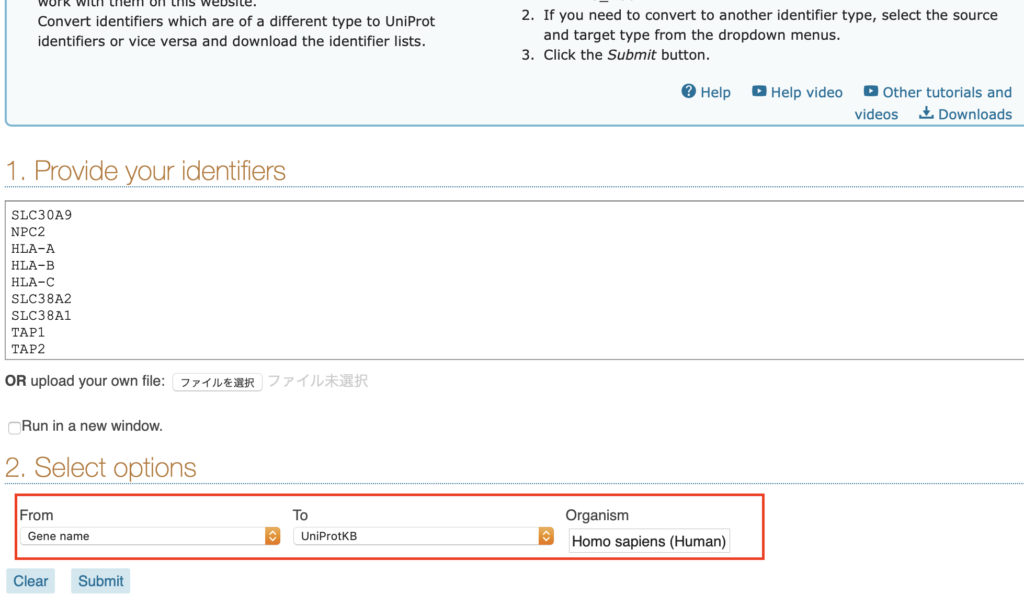
③完了したらSubmitを押してください。そうすると以下のように変換後のID候補が示されます。赤字で囲ったReviewedを押してください。これで信頼性のあるIDのみに絞られます。
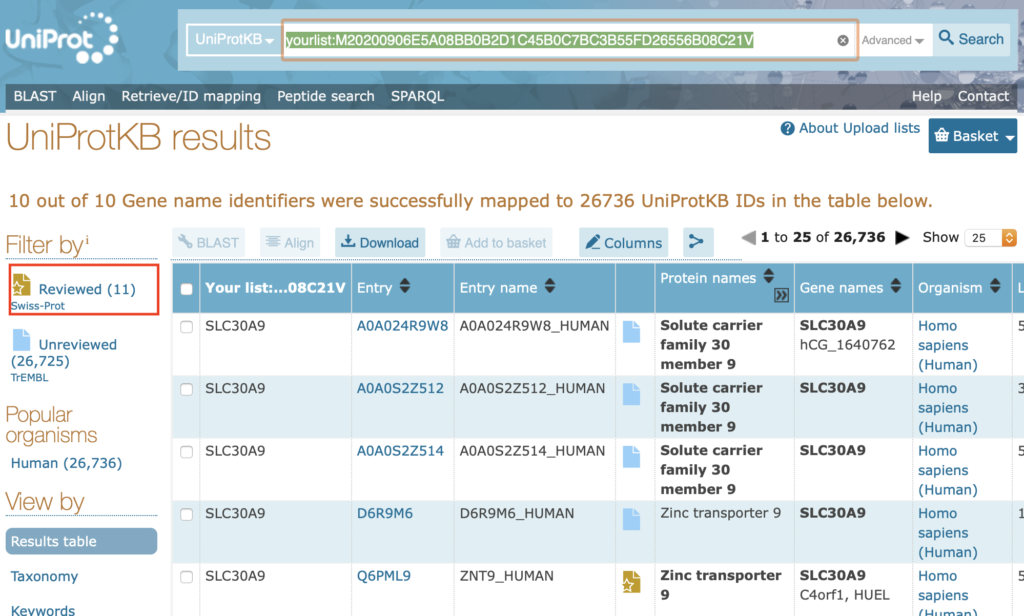
④どうやらTAP2だけ2つ候補があるようですね。どちらが正しいか、確認してください。
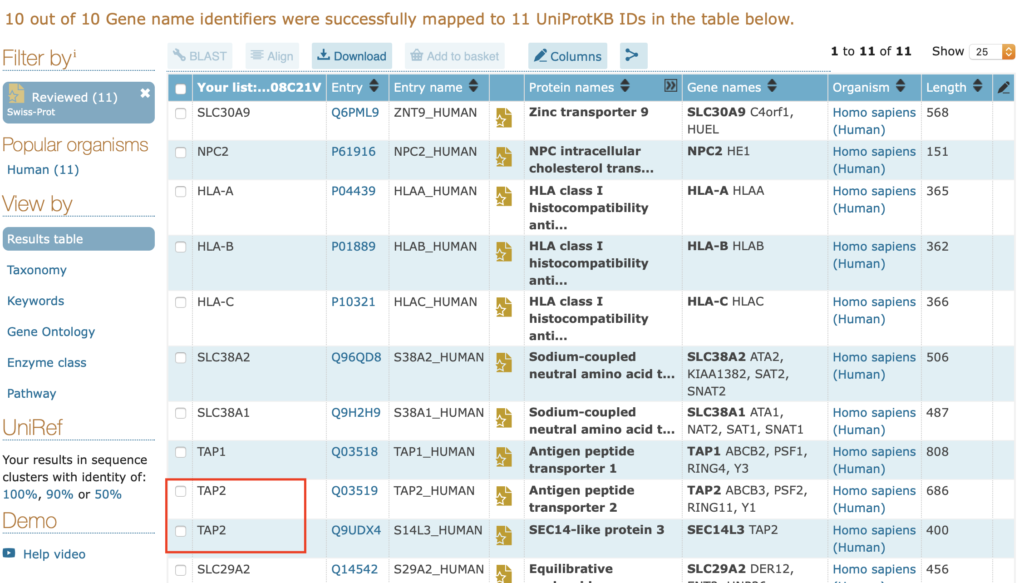
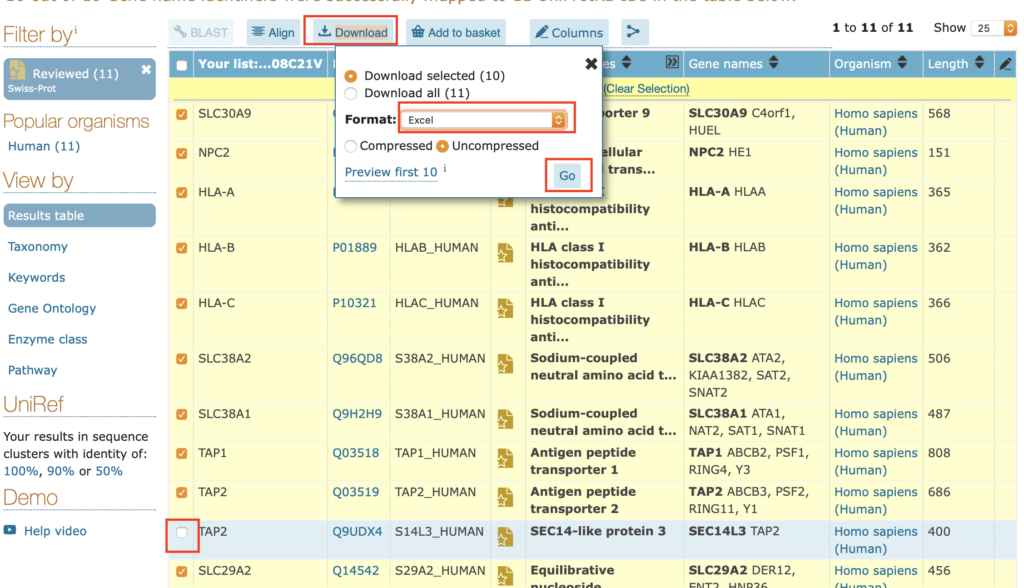
⑤ここでは全部チェックを入れた後、下のTAP2のみチェックを外して、Downloadに進みましょう。FormatはExcelでいいと思います。選択したらGoを押してください。
⑥ダウンロードしたエクセルファイルを開くと、ちゃんと変換されていますね。これをコピーして、⑦に示したように整えてください。
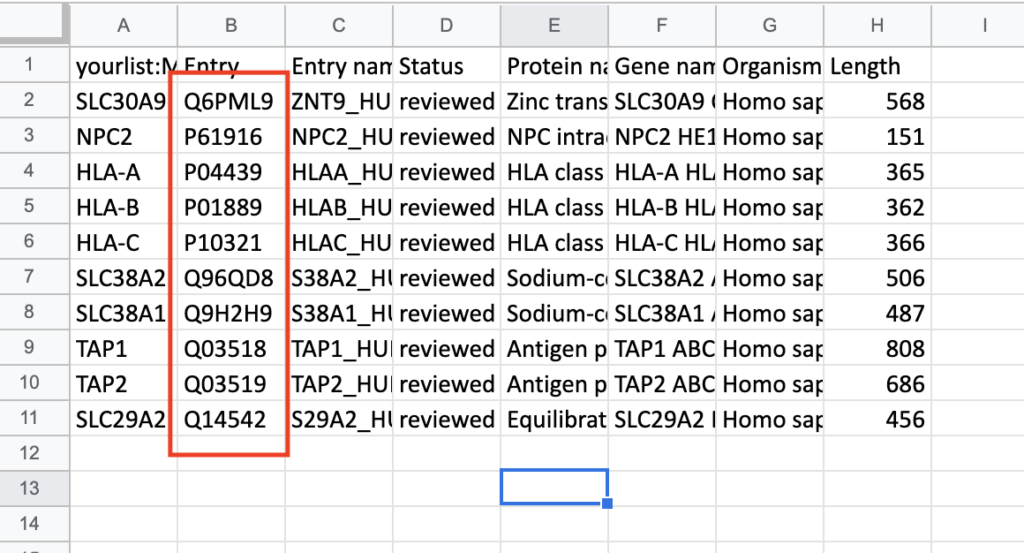
⑦IDリストの完成です。さあこれで準備が整いました!
webスクレイピングで超効率的に情報を集める
ここからはpythonを使っていきます。
pythonコード
以下のコードをpythonにコピペしましょう。これだけで各タンパク質の機能とアミノ酸配列は収集可能なのですが、応用できるよう丁寧に説明していきます。
#ライブラリのインポート
import pandas as pd
from urllib.request import urlopen
import numpy as np
from lxml import etree
#エクセルデータのインポート
#""の中にはエクセルファイルの名前と場所を入れる。
excelData = pd.read_excel("/Users/seiyaku/Download/ID_list.xlsx")
#""の中にはカラム名を入れる
uniprotIDs = excelData["uniprotKB_accession"]
#新しいカラムを作成
excelData["function"] = 0
excelData["sequence"] = 0
for uniprotID in uniprotIDs:
url = "https://www.uniprot.org/uniprot/" + uniprotID + ".xml"
f = urlopen(url)
xml = f.read()
root = etree.fromstring(xml)
#以下のコードは下の説明を参照
function = root.find('./entry/comment[@type="function"]', root.nsmap)
if function==None:
pass
else:
excelData["function"][uniprotIDs==uniprotID] = function[0].text
print(function[0].text)
sequence = root.find('./entry/sequence', root.nsmap)
if sequence==None:
pass
else:
excelData["sequence"][uniprotIDs==uniprotID] = sequence.text
print(sequence.text)
#結果をエクセルファイルとして書き出し
excelData.to_excel("uniprot_Data.xlsx")
uniprot API
今回は、uniprotのAPIというものを利用しています。公式の説明はこちらを見てみてください。
各IDには、.xmlという形式でアクセスしています。例えば、Q6PML9の.xmlにアクセスしてみましょう。

アドレスを見ると分かりますが、Q6PML9というIDに.xmlが続いています。ちなみにこの.xmlをとると、通常のuniprotの検索画面となります。
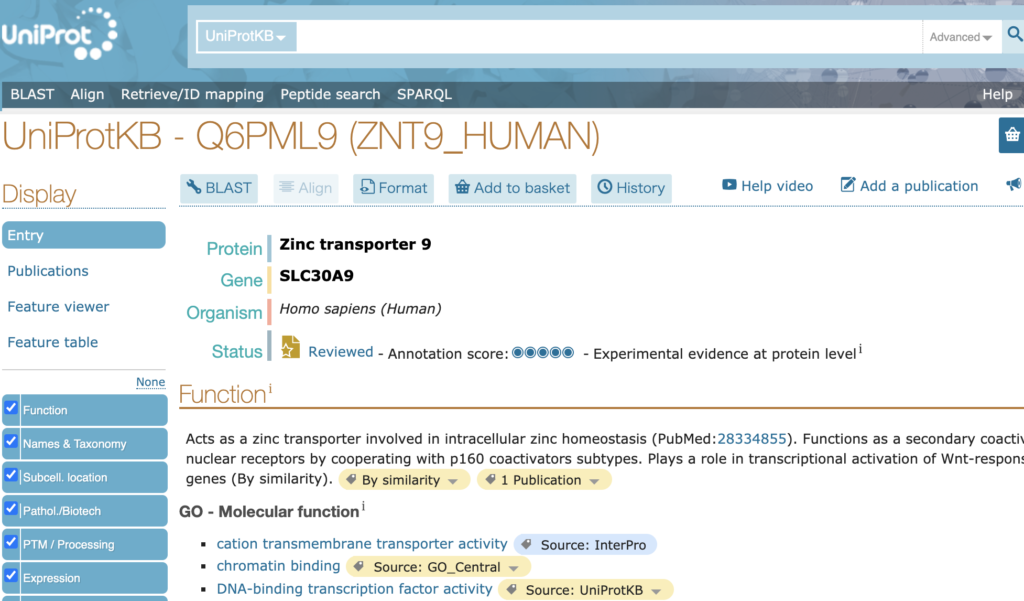
↓そしてこれが.xmlの画面です。先程のコードではタンパク質の機能とアミノ酸配列を収集しましたが、収集したい情報を変えるには、この.xmlをみてコードを変換する必要があります。
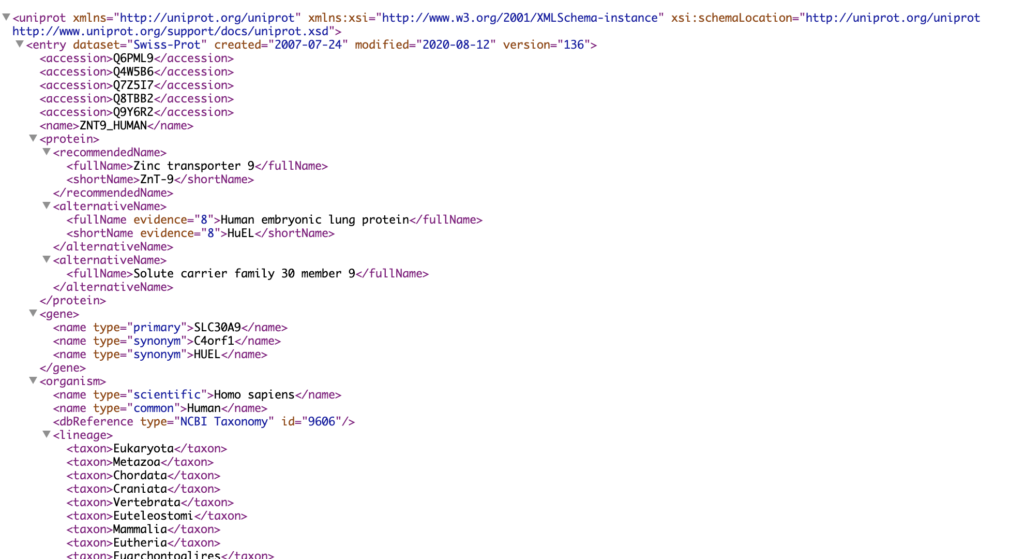
上記のコードには以下の部分があったと思いますが、このroot.findの中身を変換することで収集する情報を変換できます。
function = root.find('./entry/comment[@type="function"]', root.nsmap)
if function==None:
pass
else: excelData["function"][uniprotIDs==uniprotID] = function[0].text
print(function[0].text)
例えばZinc transporter 9という名前を取ってきたい時は、以下のコードになります。
proteinName = root.find('./entry/protein/recommendedName/fullName', root.nsmap)
if function==None:
pass
else: excelData["proteinName"][uniprotIDs==uniprotID] = proteinName.text
print(proteinName.text)
このように、例として示したコードと.xmlを見比べて、必要なデータに合わせてコードを徐々に追加してみましょう。
実際に実行すると以下のようなエクセルファイルが得られます。
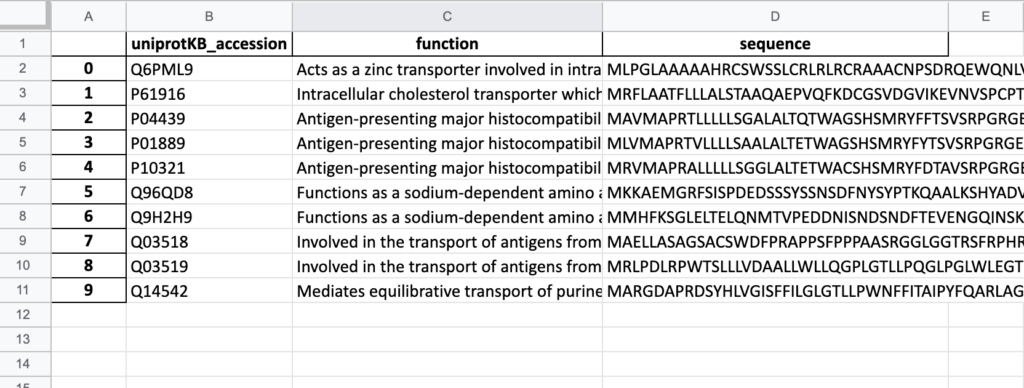
たくさんのデータを一気に集めることができました!
今回は10タンパク質分しか行いませんでしたが、100, 1000タンパク質分となってくると、この手法のありがたみがわかってくると思います。
わざわざuniprot側が用意しているものなので、使わない手は無いですね!!
エラーが起きた場合
もしコードを実行してみてエラーが起きた場合、必要なライブラリがインストールされていないかもしれません。その場合、以下のようなエラーが出ます。
![]()
そのような場合は、以下のライブラリのインストールを行ってください。
- lxml
- xlrd
- openpyxl
それでもわからない場合
この投稿にコメントしてくだされば、可能な限りお答えしたいと思います。
ぜひ試してみてください!

コメント